Статья:




СИСТЕМАТИЧЕСКИЙ ПОДХОД К СИНДРОМУ АРНОЛЬДА-КИАРИ
Секция: 4. Медицинские науки
Выходные данные
Хакимова И.Д. СИСТЕМАТИЧЕСКИЙ ПОДХОД К СИНДРОМУ АРНОЛЬДА-КИАРИ // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XXIV междунар. студ. науч.-практ. конф. № 5(23). URL: https://nauchforum.ru/archive/MNF_nature/5(23).pdf (дата обращения: 06.04.2025)
Лауреаты определены. Конференция завершена
Эта статья набрала 6 голосов
Мне нравится14
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XXIV Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
СИСТЕМАТИЧЕСКИЙ ПОДХОД К СИНДРОМУ АРНОЛЬДА-КИАРИ
Хакимова Ирина Дамировна
студент Первого московского государственного медицинского университета имени И.М. Сеченова, РФ, г. Москва
Дегтяревская Татьяна Юрьевна
научный руководитель, канд. биол. наук, доц. кафедры биологии и общей генетики Первого московского государственного медицинского университета имени И.М. Сеченова, РФ, г. Москва
Введение.
Синдром (аномалия) Арнольда-Киари — это врожденное заболевание, связанное с патологией развития ромбовидного мозга, проявляющаяся несоответствием размеров задней черепной ямки (ЗЧЯ) и мозговых структур, находящихся в этой области. Актуальной проблемой при этом синдроме является нарушение нормальной работы близлежащих мозговых структур, что при отсутствии надлежащего лечения приводит к появлению симптомов поражения продолговатого, спинного мозга и мозжечка.
Общие сведения.
Среди дизрафических расстройств особое место занимает аномалия Арнольда-Киари, которая характеризуется водянкой мозга и spina bifida. Как известно, у эмбрионов нижний конец спинного мозга располагается в крестцовом отделе позвоночника. В последующем, благодаря тому, что рост позвоночника обгоняет рост спинного мозга, conus medullaris сдвигается вверх до уровня II поясничного позвонка. Синдром Арнольда-Киари возникает в связи с тем, что закладка спинного мозга в нижнем отделе срастается с задней стенкой позвоночного канала и в процессе роста не происходит подтягивания нижнего отдела спинного мозга вверх. Напротив, продолговатый мозг и мозжечок отчасти втягиваются через большое затылочное отверстие в позвоночный канал. В крайних случаях нижняя часть продолговатого мозга обнаруживается на уровне III—IV шейных позвонков. Вследствие вклинивания мозга в большое затылочное отверстие Лушки (латеральная апертура IV желудочка) сдавливается, и ликвор может выходить их желудочковой системы только через отверстия Мажанди (срединная апертура IV желудочка). Вместе с тем, из-за сдавливания субарахноидальных пространств в окружности продолговатого мозга поступление ликвора из отверстия Мажанди к поверхности больших полушарий оказывается затрудненным, и это ведет к постепенно развивающийся гидроцефалии, которая может усугубить вклинивание продолговатого мозга и мозжечка.
Как правило, гидроцефалия при синдроме Арнольда-Киари развивается только после рождения, что может быть сопоставлено с прекращением подтекания ликвора из области spina bifida [3, с. 105]. Как правило, этот порок возникает при наличии спинномозговых грыж, чаще менингомиелоцеле, в поясничной области [Agmanolis D. et al., 1984]. Вклинение продолговатого мозга и мозжечка в большое затылочное отверстие, очевидно, происходит вследствие тракций спинного мозга, дистальный конец которого фиксирован в области грыжевого мешка. Порок Арнольда-Киари может развиться и прогрессировать на протяжении почти всего внутриутробного периода. По мере роста эмбриона и плода фиксированный каудальный конец спинного мозга втягивает мозжечок и продолговатый мозг в позвоночный канал. Это в свою очередь приводит к сдавлению указанных апертур IV желудочка, нарушению оттока спинномозговой жидкости и развитию внутренней гидроцефалии. Продолговатый мозг при этом деформирован, мозжечок отстает в развитии и наслаивается на продолговатый мозг.
Вклинение миндалин мозжечка в большое затылочное отверстие может быть следствием повышения внутричерепного давления любой другой этиологии, например, опухоли, воспаления головного мозга, гидроцефалии и др. Однако во всех таких случаях вклинение гораздо меньше, чем при пороке Арнольда-Киари.
Частота заболевания Арнольда-Киари составляет от 3.3 до 8.2 наблюдений на 100000 населения. До настоящего времени патогенез заболевания Арнольда-Киари окончательно не установлен. Ученые выявили три патогенетических фактора, приводящих к заболеванию синдромом Арнольда-Киари: наследственно обусловленные врожденные остеоневропатии, травматические повреждения клиновидно-решетчатой и клиновидно-затылочной части ската вследствие родовой травмы, гидродинамический удар ликвора в стенки центрального канала спинного мозга.
Распространённость заболевания и наследственность.
Ранние исследования предполагали, что распространенность сирингомиелии составляет 1 на 18000 (Small, Sheridan, 1966). На самом деле, эти цифры ниже реальных, потому что основываны на данных аутопсии, а не МР сканирования. Лучшим способом определить распространенность этих заболеваний, является проспективное исследование. До настоящего времени такого исследования не проводилось из-за его дороговизны и трудоемкости, к тому же, синдром Арнольда-Киари довольно «молодое» заболевание, поскольку для его точного диагностирования применяется сравнительно новая технология МРТ.
По последним данным, врачи и ученые из университета Джонса Хопкинса (США) исследовали более 22000 МР сканов головного мозга (Meadows, Kraut et al, 2000). Это самая большая серия, описанная в научном исследовании, и у 1 из 1280 индивидов была выявлена аномалия Киари I (определяемая как опущение миндалин мозжечка ниже затылочного отверстия на 5 мм или более). Можно критиковать это исследование, поскольку оно было выполнено в крупных медицинских центрах (пациенты с необычными симптомами чаще попадают в такие центры). Однако любые данные о частоте заболевания не могут быть до конца точными, поскольку синдром Арнольда-Киари первого типа может протекать бессимптомно. Соответственно пациенты с бессимптомным протеканием заболевания включены не были.
Более 15 последних лет многие ученые докладывали о случаях аномалии Арнольда-Киари и сирингомиелии у нескольких членов семей. Недавно было доказано, что расстройство имеет генетическую основу (Milhorat, Chou et al, 1999; Speer, Enterline et al, 2003).
Дальнейшие доказательства в поддержку генетической природы некоторых случаев аномалии Арнольда-Киари и сопутствующей (не всегда) сирингомиелии были получены при близнецовых исследованиях (когда у одного из однояйцевых близнецов имеется аномалия Арнольда-Киари и/или сирингомиелия, у другого близнеца это состояние возникает чаще, чем в случаях с разнояйцевыми близнецами) (Speer, Enterline et al. 2003).
Типы аномалий.
В 1891 году Киари выделил четыре типа заболевания, получивших в дальнейшем название «аномалия Арнольда-Киари» с подробным их описанием.
Аномалия Киари 1-го типа — дистопия миндалин мозжечка, т. е. опущение их в большое затылочное отверстие и позвоночный канал со сдавлением продолговатого и верхних отделов спинного мозга. При этом ствол мозга расположен обычно, черепные нервы не смещены, нет признаков нарушения их функции. Примерно в 50 % случаев сочетается с сирингомиелией (образование полостей в веществе спинного мозга), реже — с закрытой гидроцефалией.

Рисунок 1. На левом снимке представлена МР картина Аномалии Арнольда-Киари I типа — опущение миндалин мозжечка ниже линии Чемберлена (линия, проведенная от заднего края затылочного отверстия до твердого неба). Ниже места ущемления визуализируется сирингомиелическая киста. На правом снимке представлен вариант нормы
В основе аномалии Киари 1-го типа лежит нарушение динамики ликвора на уровне краниовертебрального перехода. За счет дополнительных арахноидальных мембран и спаек выходящий из отверстия Мажанди ликвор не распределяется равномерно по внутричерепному и спинальному субарахноидальным пространствам, но направляется преимущественно вверх, интракраниально. При этом пульсовая волна ликвора оказывает давление на самые нижние отделы — миндалины мозжечка. Это приводит к затруднению венозного оттока, увеличению объема миндалин, их фиброзированию и постепенному смещению вниз. Соответственно нарушения ликвородинамики на уровне краниовертебрального перехода при этом усугубляются, возникает разобщение внутричерепного и спинального ликворных пространств, и из-за градиента давления увеличивается степень смещения миндалин мозжечка. Ликворная волна в нижних отделах IV желудочка в каждую систолу оказывает давление на слепое отверстие, и примерно у 50 % больных ликвор начинает поступать в центральный канал спинного мозга, приводя к возникновению сирингомиелии. Процесс развивается очень медленно, поэтому врожденная аномалия приводит к появлению первых клинических симптомов в среднем и иногда — в старшем возрасте.
Аномалию Киари 1-го типа следует отличать от вторичного смещения миндалин мозжечка при объемных процессах, приводящих к повышению внутричерепного давления и дислокации мозжечка в большое затылочное отверстие (травма, опухоль и т. д.).
Клиническая картина. Наиболее характерны боли в шейнозатылочной области, которые могут усиливаться при сгибании головы и натуживании. Возможны нарушения координации, статики и походки, дизартрия, спонтанный нистагм. При развитии сирингомиелии появляются характерные нарушения чувствительности и движений. Иногда развиваются гидроцефалия и признаки повышения внутричерепного давления. Средний возраст появления первых симптомов — около 40 лет, немного чаще страдают женщины.
Диагностика. Стандарт диагностики — МРТ без контрастного усиления. На МР-томограммах головы в сагиттальной плоскости выявляется смещение миндалин мозжечка книзу от нижнего края большого затылочного отверстия (степень дистопии с выраженностью клинической симптоматики не коррелирует). При МРТ спинного мозга может быть выявлена сирингомиелия.

Рисунок 2. Аномалия Киари 1-го типа, сочетающаяся с сирингомиелией. МРТ, Т1-взвешенное изображение. Миндалины мозжечка опущены до С1 позвонка. Центральный канал спинного мозга резко расширен, заполнен ликвором
Дифференциальную диагностику следует проводить с вторичной дислокацией миндалин мозжечка при повышении внутричерепного давления (вследствие опухоли, гематомы и т. д.) и с другими видами аномалии Киари (см. ниже).
Лечение. Единственным эффективным способом лечения аномалии Киари 1-го типа, проявляющейся клинически, является хирургическое вмешательство. Если аномалия Киари 1-го типа является случайной находкой, осуществляется динамическое наблюдение за больным. Наибольший эффект дает операция, выполненная в первые 2 года после появления клинических симптомов.
Под наркозом производят небольшой разрез кожи в шейнозатылочной области по средней линии, раздвигают мягкие ткани и резецируют край большого затылочного отверстия и заднюю дужку Ср а при значительной дистопии миндалин — и Сп. Линейно в вертикальном направлении рассекают ТМО и производят свободную пластику образовавшегося дефекта лоскутом синтетической оболочки или фасции. На этом операция заканчивается, рану зашивают наглухо. Такое вмешательство практически безопасно и в большинстве случаев приводит к быстрому регрессу симптомов. Прогноз благоприятный.
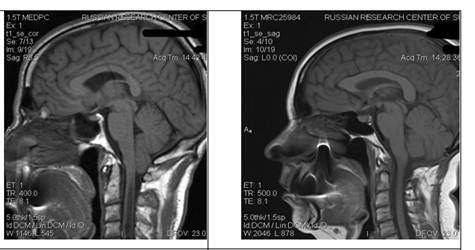
А) Б)
Рисунок 3: Результаты оперативного лечения. А) МРТ пациента с аномалией Арнольда-Киари 1 типа, сирингомиелией шейного отдела спинного мозга (миндалики мозжечка опущены на 10 мм ниже линии Чемберлена, с уровня С2 опеределяется сирингомиелическая полость). Пациента беспокоила сильная головная боль, боль в шейном отделе позвоночника, онемение в кистях, легкая слабость в руках; Б) МРТ того же больного через 6 мес. после операции (миндалики мозжечка находятся над линией Чемберлена, сирингомиелическая полость исчезла). Вся симптоматика у пациента регрессировала
Аномалия Киари 2-го типа — уродство развития, при котором весь ствол мозга (от моста до продолговатого мозга) и IV желудочек смещены каудально. Миндалины мозжечка могут занимать как нормальное положение, так и быть смещены каудально. В большинстве случаев наблюдается гидроцефалия, могут встречаться микрогирия, гипоплазия серповидного отростка, отсутствие прозрачной перегородки. Возможно развитие сирингомиелии. У большинства больных аномалия Киари 2-го типа сочетается с миеломенингоцеле (см. ниже), аномалиями развития костей черепа и позвоночника (ассимиляцией атланта, сращением шейных позвонков друг с другом, базилярной импрессией).
Клиническая картина. Для новорожденных характерны нарушения глотания, периоды апноэ, стридорозное дыхание вследствие паралича голосовых складок, аспирации, опистотонус или общая гипотония, спонтанный нистагм, слабый плач или его отсутствие, гипомимия или амимия. Если указанные симптомы выявляются сразу после рождения, прогноз плохой, дети погибают обычно в течение нескольких дней. Для детей более старшего возраста характерны в первую очередь нарушения глотания, фонации, слабость в руках. Чем больше возраст, в котором появились симптомы, тем лучше прогноз.
Диагностика. Стандарт диагностики — МРТ без контрастного усиления. На МР-томограммах верхнешейного отдела позвоночника выявляются Z-образный изгиб в области перехода смещенного каудально продолговатого мозга в спинной, гидроцефалия, костные аномалии и прочие указанные выше морфологические изменения.

Рисунок 4. Аномалия Киари 2-го типа. МРТ, Т1-взвешенное изображение. Ствол мозга и мозжечок смещены каудально, IV желудочек сдавлен на уровне краниовертебрального перехода, почти не дифференцируется, также определяются спинно-мозговая грыжа на верхнегрудном уровне и сирингомиелия (ниже). МРТ спинного мозга подтверждает диагноз миеломенингоцеле
Лечение. Новорожденных и грудных детей, как правило, не оперируют. В остальных случаях производят декомпрессию задней черепной ямки, резекцию дужек верхних шейных позвонков со свободной пластикой ТМО.
Прогноз определяется тяжестью поражения ствола головного мозга и выраженностью неврологического дефицита. В среднем хирургическое вмешательство обеспечивает улучшение или полный регресс симптомов у 2/3 оперированных больных.
Аномалия Киари 3-го типа. Смещение всех структур задней черепной ямки, включая мозжечок, каудально. Обычно сочетается с затылочным энцефаломенингоцеле или с шейным миеломенингоцеле. Способов лечения не существует. Прогноз крайне неблагоприятный, патология несовместима с жизнью. К счастью, встречается крайне редко.
Аномалия Киари 4-го типа. Гипоплазия мозжечка без дислокации. Лечения не требует.
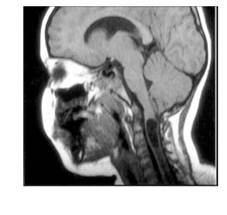
Рисунок 5. МРТ изображение (T1-взвешенное изображение) у ребенка и аномалией Арнольда-Киари 0 типа и сирингомиелией
Оперативное вмешательство.
В настоящее время общепризнанной операцией является декомпрессивная трепанация задней черепной ямки и удаление дужки первого шейного позвонка.
Цель операции – восстановить циркуляцию спинно-мозговой жидкости и, следовательно, достичь улучшения состояния пациента (за счет декомпрессии ствола головного и уменьшения сирингомиелической полости). Так же очень важно предупредить рецидив заболевания и избежать его прогрессирования в будущем.
После операций на краниовертебральном переходе, в случаях нормализации ликвородинамики и устранения диссоциации краниоспинального давления, клинические проявления сирингомиелии могут стабилизироваться или уменьшиться даже без значимых визуальных изменений сирингомиелической кисты. одной из задач операции является профилактика дальнейшего роста кист и прекращение прогрессирования сирингомиелического синдрома.
В результате хирургического лечения симптомы сирингомиелии при аномалиях краниовертебрального перехода поддаются различной степени регресса у 30—45 % больных, стабилизация процесса с остановкой прогрессирующего ухудшения отмечается у 45 %. Операция не оказывает влияния на характер течения заболевания у 10—20 % пациентов. Летальность при этом виде патологии в настоящее время не превышает 1—2 %.
При наличии положительной динамики уменьшение двигательных расстройств происходит у 25 % больных, регресс расстройств поверхностной чувствительности — у 30 % больных, парестезий — у 25 % больных. Рецидивы заболевания и дальнейшее его прогрессирование после временного улучшения или стабилизации наблюдаются у 20—40 % больных. Это может быть связано с прогрессированием рубцово-спаечного процесса в области краниовертебрального перехода, дальнейшей каудальной дислокацией мозжечка в костный дефект, развитием спаечного процесса в месте сирингошунтирующей операции, смещением шунта и т. д. Плохими клиническими прогностическими факторами при хирургическом лечении сирингомиелии считают выраженные мышечные атрофии, грубые диссоциированные чувствительные расстройства на большей части поверхности тела, выраженный сколиоз, длительный анамнез заболевания [1, c. 8].
Ход операции.
Если отсутствуют костные изменения, то наличие клинически значимой аномалии Арнольда-Киари потребует субокципитальной краниектомии (увеличение большого затылочного отверстия путем выпиливания небольшого фрагмента затылочной кости и удаления дужки первого шейного позвонка) для уменьшения прямого давления на ствол головного мозга и нормализации циркуляции спинно-мозговой жидкости. После удаления костных структур, выполняется пластика твердой мозговой оболочки (ТМО), что позволяет увеличить объем задней черепной ямки. Пластика ТМО проводится собственными тканями пациента (надкостница, апоневроз) или замещающего материала (животного или искусственного происхождения). Иногда хирург во время операции использует ультразвуковое исследование для оценки движения и пульсации спинно-мозговой жидкости, а так же для определения уровня опущения миндалин мозжечка. Ультразвуковое исследование помогает визуализировать соотношения между костями черепа, стволом головного мозга и мозжечком. В большинстве случаев, операция проводится под операционным микроскопом. Во время операции хирург принимает решение в отношении резекции (удаления) опущенных миндалин мозжечка.
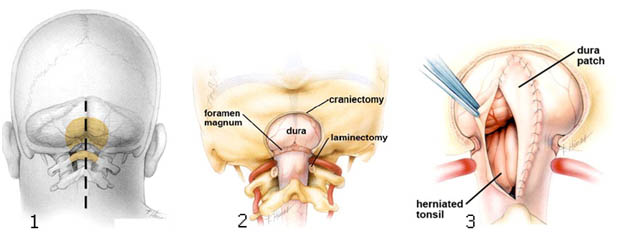
Рисунок 6. Схематическое изображение классической субокципитальной краниектомии. После того как пациент уснет под действием наркоза, его укладывают на операционный стол вниз лицом и фиксируют голову в специальном фиксаторе черепа. 1 — после обработки кожи раствором антисептика производится разрез кожи и мышц шеи по средней линии и обнажаются кости черепа и первый шейный позвонок. 2 — производится удаление небольшого участка затылочной кости (краниектомия) и дужки первого шейного позвонка (ламинектомия). 3 — вскрывается твердая мозговая оболочка, осматривается зона компрессии миндаликов мозжечка (при необходимости проводится их субпиальная резекция). Выполняется пластика твердой мозговой оболочки различными трансплантатами (вшивается «заплатка»)
После выполнения основного этапа операции хирург послойно ушивает рану. Небольшому числу пациентов может потребоваться задняя стабилизация титановыми пластинами. Установка шунта остается на усмотрение лечащего врача в каждой конкретной ситуации. Обычно, операция длится 2—4 часа.
Заключение.
Синдром Арнольда-Киари на сегодняшний день относится к тяжелым врожденным заболеваниям нервной системы, трудно диагностируемом в раннем возрасте. Но благодаря современному хирургическому лечению таких о пациентов устраняются симптомы поражения мозжечка, продолговатого и спинного мозга. Однако генетическая природа заболевания изучена не до конца. Именно поэтому очень важно понимать механизм возникновения данного заболевания, чтобы уметь не только устранить, но и предотвратить нежелательные последствия данного заболевания.
Список литературы:
1. Евзиков Г.Ю. Сирингомиелия // Нейрохирургия. — 2008. — № 2. — С. 8—13.
2. Неврология и нейрохирургия. Гусев Е.И., Коновалов А.Н., Бурд Г.С. «Медицина», 2000.
3. Невропатология раннего детского возраста. Руководство для врачей. Б.В. Лебедев, Ю.И. Барашнев, Ю.А. Якунин. Издательство «Медицина», Москва, 1981.
4. Тератология человека. Руководство для врачей / Кириллова И.А., Кравцова Г.И., Кручинский Г.В. и др.; Под ред. Г.И. Лазюка. — 2-е изд., перераб. и доп. — Ж: Медицина, 1991.
