Статья:




СИНТЕЗ ГЕРБИЦИДОВ НА ОСНОВЕ СУЛЬФОНИЛМОЧЕВИН
Секция: 1. Химические науки
Выходные данные
Волков В.М. СИНТЕЗ ГЕРБИЦИДОВ НА ОСНОВЕ СУЛЬФОНИЛМОЧЕВИН // Молодежный научный форум: Естественные и медицинские науки: электр. сб. ст. по мат. XIII междунар. студ. науч.-практ. конф. № 6(13). URL: https://nauchforum.ru/archive/MNF_nature/6(13).pdf (дата обращения: 26.04.2025)
Лауреаты определены. Конференция завершена
Эта статья набрала 0 голосов
Мне нравится0
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников
Дипломы
лауреатов
лауреатов
Сертификаты
участников
участников

XIII Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки»
СИНТЕЗ ГЕРБИЦИДОВ НА ОСНОВЕ СУЛЬФОНИЛМОЧЕВИН
Волков Вячеслав Михайлович*
студент з/о, ФГБОУ ВПО «МАМИ», РФ, г. Москва
Антонова-Антипова Ирина Петровна
научный руководитель, канд. хим. наук, проф., ФГБОУ ВПО «МАМИ», РФ, г. Москва
Федосеевский Владимир Викторович
Иванова Елена Павловна
Метсульфурон-метил — гербицид системного действия класса сульфонил-мочевин, проникающий через корни и листья растений и ингибирующий биосинтез изолейцина и валина, впервые был получен и заявлен фирмой Дюпон в США в 1986 г.:
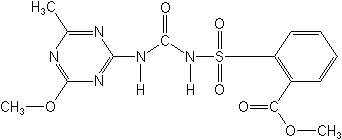
В настоящее время метсульфурон-метил широко используется в сельском хозяйстве для борьбы с однолетними двудольными сорняками на посевах зерновых колосовых культур (пшеница яровая и озимая, ячмень, овес) [2].
Обзор посвящен анализу литературы по способам получения гербицидов класса сульфонилмочевин, а также соединений, используемых для их синтеза.
Все гербициды класса сульфонилмочевин содержат так называемый сульфонилмочевинный мостик -SO2NHCONH-, связанный с ароматическим циклом и гетероциклом. Таким образом, гербициды сульфонилмочевины — соединения общей структуры R1-SO2NHCONH-R2, где R1 — арил или алкил, ___________________
*Волков В.М. — ФБУН «Федеральный научный центр гигиены им. Ф.Ф.Эрисмана» РОСПОТРЕБНАДЗОРА
R2 — гетероциклический фрагмент, чаще всего замещенный пиримидиновый или симм-триазиновый остаток. Биологическая активность этих соединений существенным образом зависит от типов и положения заместителей в ароматическом ядре и гетероцикле [3]. Наиболее активными являются соединения триазинового ряда, содержащие атомы Cl, CF3 и некоторые другие группировки. Введение в триазиновый фрагмент молекулы гидрофильных заместителей ( -COOH, -OH) снижает активность. Введение же гидрофобных группировок (метильных, метоксильных) повышает активность этих соединений.
Важнейший метод получения сульфонилмочевин любого строения основан на реакции сульфонилизоцианатов с производными 2-аминотриазина или 2-аминопиримидина.
ArSO2N=C=O + RNH2 → ArSO2NHCONHR,
где: R — замещенный триазиновый или пиримидиновый фрагменты.
Реакция протекает как в органических растворителях, так и без них. Температура процесса зависит от строения исходных сульфонилизоцианата и амина. В качестве катализаторов можно использовать третичные амины или органические соединения олова. Полученную сульфонилмочевину отделяют кристаллизацией после удаления части растворителя. При правильном проведении процесса продукт получается практически с количественным выходом [3].
Синтез сульфонилмочевин также можно проводить ацилированием замещенных мочевин арилсульфохлоридами по следующей схеме [4]:
ArSO2Cl + RNHCONH2 → ArSO2NHCONHR
В патентах [7—14] приведено ещё 4 способа получения сульфонилмочевин. Условия реакций подробно рассмотрены в [13—14].
Способ 1. ArSO2N=C=O + RNH2 → ArSO2NHCONHR,
где: R — замещенный триазиновый (или пиримидиновый) фрагмент.
В этой реакции используют эквимолярные количества реагентов. Реакцию проводят в растворителе. В качестве растворителя используют: ароматические углеводороды (бензол, толуол); галогенированные алканы (дихлорметан, хлороформ, четыреххлористый углерод); простые эфиры (диэтиловый эфир, диизопропиловый эфир, диоксан или тетрагидрофуран (ТГФ); нитрилы (ацетонитрил); кетоны (ацетон, метилэтилкетон); ацетаты, а также диметилформамид или диметилсульфоксид.
Для ускорения реакции добавляют основания органические [триэтиламин; три(н-пропил)амин; пиридин; 1,8 –диазабицикло [5; 4; 0] — 7-ундецен (DBU); 1,4 — диазабицикло [2; 2; 2]октан (DBO); 1,5-диазабицикло- [4; 3; 0]нон-5-ен (DBN) или неорганические( гидроксид калия, гидрид натрия, карбонат калия, карбонат натрия или гидроксид натрия). Основание может быть использовано в количестве от 0,01 до 3 молей на 1 моль сульфонилизоцианата.
Продукт — сульфонилмочевину — выделяют из реакционной смеси путем отгонки растворителей или непосредственно фильтрованием. Оставшийся остаток промывают водой или разбавленной кислотой для удаления основных примесей. Кроме того, можно также растворить остаток в растворителе, не смешивающимся с водой, и промыть водой. Целевые соединения выпадают при этом в чистом виде и могут быть очищены путем перекристаллизации, перемешивания в органическом растворителе, поглощающем примеси, или хроматографически на колонке с АУ.
Данную реакцию проводят предпочтительно в ацетонитриле, метил-трет-бутиловом эфире, толуоле или метиленхлориде в присутствии от 0 до 100 молярных эквивалентов (лучше от 0 до 50).
Способ 2. ArSO2NH2 + ClCOOR1 → ArSO2NHCOOR1
ArSO2NHCOOR1 + RNH2 → ArSO2NHCONHR + R1OH,
где: R — 2-амино-1,3,5-триазин( или пиримидин), R1 — алкил(или арил).
Для ускорения реакции и улучшения качество продукта могут быть добавлены основания (триэтиламин или 1,4-диазабицикло [2; 2; 2] октан) в количестве от 0,01 до 1 моль на сульфонилкарбамат.
В качестве растворителей целесообразно применять растворители, указанные в способе 1.
Конечный продукт — сульфонилмочевину — выделяют из реакционной смеси обычным путем, как указано в способе 1.
Способ 3. ArSO2NH2 + R1OCONHR → ArSO2NHCONHR,
где: R — остаток триазина (или пиримидина), R1 — алкил или арил.
Сульфонамид ArSO2NH2 реагирует со стехиометрическим количеством гетероциклического карбамата R1OCONHR в инертном органическом растворителе при температуре 20—1000C. Для ускорения реакции и улучшения качества продукта можно добавить 0,01—1 моль основания (триэтиламин, DBO, DBU) на каждый моль исходного гетероциклического карбамата. Кроме того можно использовать неорганические основания (гидроксид калия, гидроксид натрия, карбонат калия, карбонат натрия, карбонат кальция) в количестве от 0,8 до 5 молей, обычно от 1,0 до 3,0 моль на 1 моль гетероциклического карбамата.
В качестве растворителей целесообразно применять хлороформ, ацетонитрил, диоксан от 200 до 700 мас. относительно исходного карбамата. Температура реакции может быть от -10 до 150 0C, обычно от 20 до 1000C. Продолжительность реакции от 30 минут до нескольких дней, и ее завершение может быть подтверждено с помощью тонкослойной хроматографии или высокоэффективной жидкостной хроматографии. Сульфонилмочевины выделяют из реакционной смеси методами, описанными в способе 1.
Способ 4. Сульфонамид ArSO2NH2 реагирует с изоцианатным производным триазина или пиримидина O=C=NR в ацетоне, ацетонитриле или диоксане, в присутствии карбоната калия (триэтиламина или DBU), с образованием сульфонилмочевины:
ArSO2NH2 + O=C=NR → ArSO2NHCONHR,
где: R — остаток триазина (или пиримидина).
Целевой продукт выделяют из реакционной смеси обычным путем, как описывается в способе 1.
В патенте [15] рассматривают получение галогенсодержащего аналога метсульфурон-метила — алкил 6- фтор -2-N-[[(4-метил-6-метокси-1,3,5-триазин-2-ил) аминокарбонил]амино-сульфонил]бензоата:
· реакцией метил 6-фтор-2-изоцианатосульфонилбензоата с замещенным 2-аминотриазином

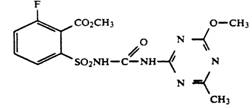
· взаимодействием 6-фтор-2-аминосульфонилбензоата с фенил-N-(4-метокси-6-метил-1,3,5- триазин-2-ил)карбаматом

· реакцией метил 6-фтор-2-(феноксикарбониламиносульфонил)бензоата с 2-амино-4-метокси-6-метил-1,3,5-триазином


В патенте [16] приведены способы получения сульфонилмочевин, в ароматическом ядре которых имеется 2 заместителя: иод- и алкоксикарбонильная группа, причем заместитель алкоксикарбонил находится как в орто-, так и в мета-положении к сульфонил-группе.
В патенте [17] подробно рассмотрены пути синтеза ортоалкокси-карбонилзамещенных сульфонилмочевин, а также сульфонилизоцианатов и сульфамидов.
Реакция арилсульфонилизоцианатов ArSO2N=C=O с аминотриазином RNH2 (R — остаток триазина или пиримидина) протекает в инертном апротонном органическом растворителе(дихлорметан, 1,2-дихлорэтан, тетрагидрофуран или ацетонитрил) при температуре от 200С до 850С.
Растворимые продукты выделяют упариванием раствора и затем их очищают растиранием остатка после упаривания с таким растворителем, как 1-хлорбутан или диэтиловый эфир, и фильтрацией, а затем перекристаллизацией из смеси растворителей, таких, как 1,2-дихлорэтан, 1-хлорбутан и гептан или хроматографией на колонке с силикагеле.
Сульфонилмочевины могут быть получены реакцией фенил-N-(арилсульфонил)карбаматов с аминогетероциклом в инертном органическом растворителе (диоксан или тетрагидрофуран) при температуре 20—1000С в течение 0,5—24 часа.
Сульфонилмочевины могут быть получены реакцией эквимолярных количеств сульфамида с соответствующим гетероциклическим фенилкарбаматом в присутствии основания (DBU):
ArSO2NH2 + C6H5OCONHR → ArSO2NHCONHR,
где: R — остаток триазина или пиримидина.
Наибольшее число патентов рассматривает синтез сульфонилмочевин через арилсульфонилизоцианаты [18—25]. Реакцию аминогетероцикла с арилсульфонилизоцианатом проводят в инертном растворителе (безводный дихлорметан).
Целью изобретения [26] является повышение выхода и качества целевого продукта — сульфонилмочевины, что достигается использованием фосгена. Фосгенирование арилсульфонамида проводят в присутствии трехкомпонентной каталитической системы, состоящей из первичного и третичного алифатических аминов и диэтилэтаноламина в качестве третьего компонента при мольном соотношении реагентов арилсульфонамид: первичный амин: третичный амин: диэтилэтаноламин = 1:0,015-0,017: 0,012-0,014:0,015-0,017, в три ступени, первоначально при 15—250C в присутствии диэтилэтаноламина, затем после догрузки первичного и третичного аминов при 60—700C и 125— 1350C до окончания реакции.
Отличительными признаками предлагаемого способа является состав катализатора, последовательность загрузки компонентов катализатора и трехступенчатый характер процесса фосгенирования арилсульфонамида.
В патентах [27—29] в качестве других методов синтеза сульфонилмочевин используют:
· реакции фенил (N-арилсульфонил)карбаматов с замещенными 2-амино-1,3,5-триазинами или пиримидинами;
· реакции арилсульфамидов с замещенными 2-фенилоксикарбониламино-1,3,5-триазинами или пиримидинами.
Согласно [30] возможно несколько вариантов для проведения
процесса. В первом варианте свободный арилсульфонамид сначала преобразуется в соль щелочного металла, например соль натрия или калия реакцией с алкоголятом натрия или калия в спирте, или в соль третичного амина. Полученную соль щелочного металла сульфонамида после сушки превращают в суспензию в апротонном растворителе, например, ацетонитриле, тетрагидрофуране, диоксане, 1,2-диметоксиэтане, диметилформамиде, N,N-диметилацетамиде или N-метилпирролидоне, а также смеси этих растворителей. Особенно подходит в качестве растворителя ацетонитрил.
Третичные аммониевые соли сульфонамидов могут быть получены в виде растворов путем смешения эквивалентного количества сульфонамида и третичного амина (DBU или DBN) в апротонном растворителе.
Полученные таким образом суспензия соли щелочного металла сульфонамида или раствор соли сульфонамида с третичным амином могут быть преобразованы в соответствующие сульфонилмочевины.
В патенте [31] приводится синтез сульфонилмочевин из замещенных пиридинсульфонамидов и N-(4-метил-6-метокси-1,3,5-триазин-2-ил)- фенил-карбамата.
В патенте [32] рассмотрены способы получения сульфонилмочевин, близких по строению к метсульфурон-метилу - алкил 2- [[(4-алкокси-6-трифторметил-1,3,5-триазин-2-ил)аминокарбонил] аминосульфонил]бензоатов:
· из алкил (2-изоцианатосульфонил) бензоатов и 2-амино-4 алкокси-6-трифторметил-1,3,5-триазинов;
· из алкил (2-аминосульфонил)бензоатов и 2-изоцианато-4-алкокси-6-трифторметил-1,3,5-триазинов;
· из алкил (2-аминосульфонил) бензоатов и 2-алкокси-4-трифторметил-6-(феноксикарбониламино)-1,3,5-триазинов.
В обзоре [5] для 2-карбоалкоксисульфонилмочевин приведен метод синтеза из соответствующего арилсульфохорида, условия реакции рассмотрены в патенте [33]:

Методы синтеза арилсульфонилизоцианатов и замещенных2-изоцианато-
1,3,5-триазинов и 2-изоцианатопиримидиновВ литературе описано несколько способов получения изоцианатов. Наиболее распространенный метод синтеза — реакция аминов с фосгеном (фосгенирование аминов) идет в среде инертного растворителя через промежуточное образование карбамонилхлоридов [1]:
RNH2 + COCl2 → RNHCOCl + HCl
RNHCOCl → RNCO + HCl
Этот метод является и основным промышленным методом получения изоцианатов.
Другой метод синтеза изоцианатов, использующийся в промышленности — термическое разложение карбаматов:
RNHCOOR' → RNCO + R'OH
В лабораторной практике вместо фосгена может быть использован оксалилхлорид, который реагирует с аминами и амидами с образованием соответствующих изоцианатов, отщепляя окись углерода [33]:
RNH2 + (COCl)2 → RNCO + СО + 2HCl
Согласно патенту [34] при получении органических изоцианатов соответствующий амин взаимодействует с фосгеном, при этом образуется органический изоцианат и хлористый водород как побочный продукт. Как правило, используется избыток фосгена таким образом, что образующийся в качестве побочного продукта газ представляет собой смесь хлористого водорода и фосгена.
Известен двухступенчатый способ получения органических изоцианатов, при котором первичные амины взаимодействуют при температурах до 80oC с избытком фосгена, и продукт, содержащий хлорангидрид карбаминовой кислоты, обрабатывают фосгеном при повышенных температурах для образования соответствующего изоцианата. Обычно взаимодействие аминов с фосгеном в промышленном масштабе осуществляется в башнях для фосгенирования (без давления или при среднем давлении) [34].
RNH2 + COCl2 → RNHCOCl + HCl
RNHCOCl → RNCO + HCl
Фосгенирование может быть проведено непрерывно с очень незначительными затратами в одну стадию, если изоцианат используется как растворитель для фосгена и эта смесь подается при комнатной температуре для фосгенирования в непрерывно функционирующие реакторы и если амин подается в чистом виде или в растворе с хорошим перемешиванием компонентов.
Арилсульфонилкарбаматы можно получать взаимодействием арилсульфонилхлоридов с алкилкарбаматами [12]:
ArSO2Cl + NH2COOR → ArSO2NHCOOR + HCl, где R = Alk.
Реакцию обычно, проводят с использованием эквимолярных количеств арилсульфонилхлорида и алкилкарбамата в растворителе в присутствии основания.
Фенилкарбаматы общей формулы C6H5OCONHR, где R — замещенный триазиновый или пиримидиновый фрагмент, могут быть синтезированы путем обработки соответствующего гетероциклического амина формулы RNH2 дифенилкарбонатом или фенилхлорформиатом в присутствии основания (гидрида натрия или пиридина) [12; 35]:
RNH2 + (C6H5O)2C=O → C6H5OCONHR или
RNH2 + C6H5OCOCl → C6H5OCONHR,
где: R — замещенный триазиновый или пиримидиновый фрагмент.
Методы синтеза 2-амино-4-метил-6-метокси-1,3,5-триазина
В [6; 36] приводятся следующие схемы синтеза 2-амино-4-метил-6-метокси-1,3,5-триазина: 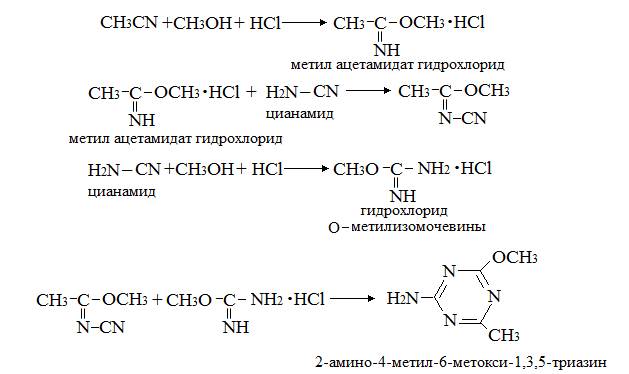
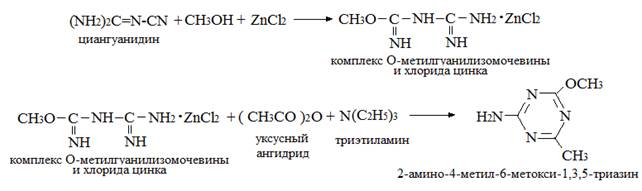
При использовании вместо уксусного ангидрида хлористого ацетила выход продукта
понижается до 60,11 % [36].
Патент [37] рассматривает получение 2-амино-4-метил-6-метокси-1,3,5- триазина реакцией
диметил N-цианоимидокарбоната (CH3O)2C=N-CN с ацетамидин гидрохлоридом CH3C(NH)NH2·HCl
или с О-метилацетамидатом гидрохлоридом CH3C(NH)OCH3·HCl в присутствии основания. 
При замене метилата натрия на КОН выход составлял 84 %. 
Выход продукта составил 43 % [37].
Замещенные 2-амино-1,3,5-триазины высокой чистоты можно легко получить с выходом
45 % реакцией N-цианоимидата R-C(N-CN)OR'' с амидином R'-C(NH)-NH2 в инертной реак-
ционной среде при температуре от 0 до 500С, предпочтительно от 20 до 300С в соответствии
со следующим уравнением [38]: 
Методы синтеза метсульфурон-метила
Наиболее биологически активным продуктом, обладающим системным воздействием на корни и листья однолетних сорняков, является метсульфурон-метил, схемы 2-х путей синтеза, которого приведены в Pesticide Synthesis Handbook [6] без описания условий реакций:
1) из 2-метоксикарбонилбензолсульфамида и этилхлорформиата в присутствии Na2CO3 получают N-(2-метоксикарбонилфенилсульфонил)этилкарбамат,
при реакции, которого с 2-амино-4-метил-6-метокси-1,3,5-триазином образуется метсульфурон-метил:

2) из 2-метоксикарбонилбензолсульфамида фосгенированием получают 2-(изоцианатосульфонил)бензоат, который, реагируя с 2-амино-4-метил -6-метокси-1,3,5-триазином, дает метсульфурон-метил:
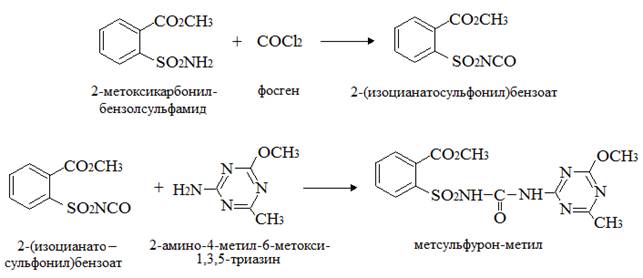 Условия получения сульфонилмочевин и, в частности метсульфурон-метила, реакцией 2-(изоцианатосульфонил) бензоата с 2-амино-4-метокси-6-метил-1,3,5-триазином, описывают патенты [19, 21] .
Условия получения сульфонилмочевин и, в частности метсульфурон-метила, реакцией 2-(изоцианатосульфонил) бензоата с 2-амино-4-метокси-6-метил-1,3,5-триазином, описывают патенты [19, 21] .
ЗАКЛЮЧЕНИЕ.
Исходя из анализа литературных данных, наиболее перспективным и экологичным методом получения метсульфурон-метила с высоким выходом является двустадийный синтез, включающий последовательные превращения по следующим схемам:
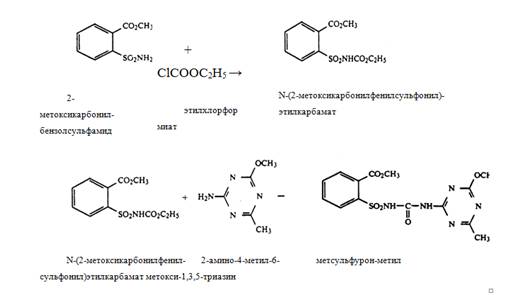
Метод позволяет исключить применение особо опасного газа — фосгена.Список литературы:
1. Горбатенко В.И., Журавлев У.З., Самарай Л.И. Изоцианаты. Методы синтеза и физико-химические свойства алкил-, арил- и гетерилизоцианатов. Киев: Наукова Думка, 1987. 447 с.
2. Захаренко В.А. Гербициды. М.: Агропромиздат, 1990. 240 с.
3. Кудрявец Ю.А., Леонтьев В.Н., Ахрамович Т.И., Сорока С.В. Биологически активные соединения в ряду сульфонилмочевины. Труды БГУ, т. 5, часть 1. 2010. С. 236.
4. Мельников Н.Н. Пестициды. Химия, технология, применение. М.: Химия, 1987. 712 с.
5. Gee S.K., Hay J.V.. Recent Developments in the Chemistry of Sulfonylurea Herbicides / Chemistry of Plant Protection. Vol. 10, 1994. P. 16.
6. Unger T. Pesticide Synthesis Handbook, 1st Edition. — 1997. — P. 185.
7. Патент RU 2088583 С1, 27.08.1997.
Patent US 4.944.793, Jul. 31, 1990.
8. Patent US 4.744.814, May 17, 1988.
9. Patent US 4.443.243, Apr. 17, 1984.
10. Patent US 5.017.212, May 21, 1991.
11. Patent EP 0238070 A2, Aug. 23, 1987
12. Patent EP 0023807 A2, Okt. 03, 1984
13. Патент РФ 2097380, 27.11.1997
14. Patent US 5.104.441, Nov. 28, 1990.
15. Патент РФ 2314291, 10.01.2008
16. Patent EP 0165003 A2, Dec. 18, 1985.
17. Patent US 4.127.405, Nov. 28, 1978
18. Patent US 4.383.113, May 10, 1983.
19. Patent US 4.420.325, Dec. 13, 1983.
20. Patent US 3.394.506, Nov. 30, 1979.
21. Patent EP 0023422 B1, Aug. 08, 1984.
22. Patent US 4.302.241, Nov. 24, 1981.
23. Patent EP 0007687 B1, March 30, 1983.
24. Patent US 4.169.719, Oct. 02, 1979.
25. Патент RU 2103263, 27.06.2008.
26. Patent EP 0237292 А2, Sept. 16, 1987.
27. Patent EP 0314505 А2, May 03, 1989.
28. Patent EP 0044807 A2, Jan. 27, 1982.
29. Patent US 4.656.273, Apr. 07, 1987.
30. Patent US 4.643.760, Feb. 17, 1987.
31. Patent EP 0388873 B1, May. 11, 1994.
32. Patent US 5.157.119, Oct. 20, 1992.
33. Патент РФ 2162840, 16.06.1995.
34. Patent US 4.699.649, Oct. 13, 1987.
35. Патент RU 2096408, 20.11.1997.
36. Patent US 5.070.199, Dec. 3, 1991.
37. Patent US 3.154.547, Oct. 27, 1964.
